

Long-QT-Syndrom: Ursachen, Diagnose, Werte, Behandlung, Medikamente
Das Long-QT-Syndrom (LQTS) bezieht sich auf eine Reihe von Symptomen, die durch eine Herzanomalie verursacht werden, die durch eine verzögerte Repolarisation von Myokardzellen gekennzeichnet ist und mit einer Synkope (Ohnmacht mit Bewusstseinsverlust und Haltungstonus) einhergeht.
Ursache der Synkope sind meist bösartige Herzrhythmusstörungen, insbesondere Spitzentorsionen, die in Kammerflimmern ausarten können und zum irreversiblen Herzstillstand des Betroffenen führen (plötzlicher Herztod).
Arrhythmien bei LQTS-Patienten werden oft durch körperliche Anstrengung oder sehr starke emotionale Reize wie Angst ausgelöst.
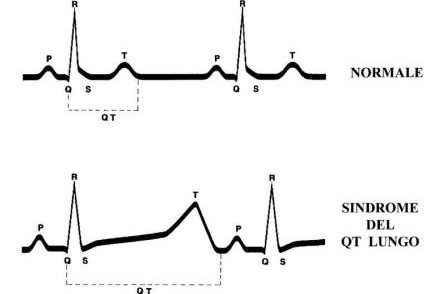
Personen mit LQTS haben eine charakteristische Verlängerung des QT-Intervalls im Elektrokardiogramm: Dieses Intervall wird vom Beginn der Q-Welle bis zum Ende der T-Welle gemessen (siehe Abbildung oben).
Allen Formen von LQTS liegt eine abnormale Repolarisation des Myokards zugrunde.
Anomalien in der Repolarisation verursachen Unterschiede in der myokardialen Refraktärität.
Aufgrund dieser Unterschiede können sich Nachdepolarisationen (die bei LQTS-Patienten häufiger auftreten) auf benachbarte Zellen ausbreiten und zu ventrikulären Reentry-Arrhythmien führen.
Epidemiologie und Risikofaktoren beim Long-QT-Syndrom (LQTS)
Bei genetisch prädisponierten Personen sind plötzliche Anstiege des sympathischen Tonus, wie sie bei übermäßiger Anstrengung oder heftigen Emotionen auftreten können, Risikofaktoren für das Auftreten maligner Arrhythmien.
Das Syndrom betrifft überwiegend junge Frauen. Bei den Betroffenen ist die Inzidenz von Spitzentorsion, Synkope und plötzlichem Herztod höher bei angeborener Taubheit, vorausgegangener Tachyarrhythmie oder Synkope; er nimmt auch proportional zur Verlängerung des QT-Intervalls zu.
Das relative Risiko maligner Arrhythmien erhöht sich schätzungsweise um das 1.1- bis 1.2-fache für jede Verlängerung des QTc-Intervalls um 10 ms über die normalen Werte hinaus.
Klinische Varianten
Wir unterscheiden zwei verschiedene angeborene Syndrome, die durch eine Verlängerung des QT-Intervalls und das Risiko eines plötzlichen Todes durch ventrikuläre Arrhythmien gekennzeichnet sind:
- Romano-Ward-Syndrom, das nach dem autosomal dominanten Modell vererbt wird (nicht assoziiert mit angeborener neurologischer Taubheit oder anderen angeborenen Herzerkrankungen, Autismus, vollständiger Syndaktylie und Immunschwäche).
- Jervell-Lange-Nielsen-Syndrom, das autosomal-rezessiv vererbt wird (assoziiert mit angeborener neurologischer Taubheit oder anderen angeborenen Herzerkrankungen, Autismus, kompletter Syndaktylie und Immunschwäche).
Ursachen von LQTS
Es gibt zwei Formen von LQTS: angeboren (seltener) und erworben (häufiger).
Erworbene Formen
Die meisten in der klinischen Praxis beobachteten Fälle betreffen erworbene Formen, die in zwei Kategorien unterteilt werden können: solche, die durch Störungen des Hydroelektrolythaushalts verursacht werden, und solche, die von der Verabreichung von Medikamenten abhängig sind.
Durch Elektrolytstörungen induzierte Formen
- Hypokaliämie
- Hypomagnesiämie
- Hypokalzämie
- Drogeninduzierte Formen
- Antiarrhythmika
- Quinidine
- Amiodaron
- Sotalol
- Procainamid
- Ranolazin
- Antihistaminika
- Terfenadin
- Astemizol
- Mizolastin
- Makrolid-Antibiotika
- Erythromycin
- Bestimmte Fluorchinolon-Antibiotika
- Wichtige Anxiolytika
- Trizyklische Antidepressiva
- Wirkstoffe, die auf die Magen-Darm-Motilität wirken
- Cisaprid
- Domperidone
- Antipsychotika
- Haloperidol
- Quetiapin
- Thioridazine
- Droperidol
- Analgetika
- Methadon
- ich LAAM
Ebenso wie angeborene Formen des LQTS können erworbene Formen zu lebensbedrohlichen Herzrhythmusstörungen führen.
Die Behandlung besteht aus der Korrektur des Elektrolytungleichgewichts, der Beseitigung seiner Ursache und dem Absetzen der Therapie mit dem Medikament, das auf eine QT-Verlängerung hinweist.
Angesichts seiner breiten Anwendung, der Neigung zu Wechselwirkungen mit anderen Arzneimitteln und der inhärenten Fähigkeit, eine Verlängerung des QT-Intervalls zu verursachen, ist Erythromycin wahrscheinlich die vorherrschende Ursache des erworbenen Long-QT-Syndroms.
Tatsächlich ist die Anwendung von Erythromycin mit einer mehr als doppelt so hohen Inzidenz plötzlicher Todesfälle verbunden wie bei anderen Antibiotika.
Zusätzlich zu den beiden oben aufgeführten Hauptkategorien muss daran erinnert werden, dass es andere Ursachen für eine Verlängerung des QT-Intervalls gibt, wie z. B. Anorexia nervosa, Hypothyreose, HIV-Infektion, Myokarditis und Myokardinfarkt.
Angeborene Formen
Angeborene Formen von LQTS können durch Mutationen in einem von mehreren bisher identifizierten Genen bestimmt werden.
Diese Mutationen neigen dazu, die Dauer des ventrikulären Aktionspotentials (APD) zu verlängern und somit das QT-Intervall zu verlängern.
Die angeborenen Formen können autosomal-dominant oder autosomal-rezessiv vererbt werden.
Die autosomal-rezessive Form ist assoziiert mit anderen angeborenen Herzerkrankungen, Autismus, kompletter Syndaktylie und Immunschwäche (LQTS8) oder mit angeborener neurologischer Taubheit (LQTS1).
Im Zusammenhang mit LQTS werden immer mehr Genorte identifiziert. Gentests für LQTS sind in der klinischen Praxis verfügbar und können auch bei der Festlegung der geeigneten Therapie helfen (Übersicht über LQTS-Gentests).
Die häufigsten Ursachen für LQTS sind Mutationen in den Genen KCNQ1 (LQTS1), KCNH2 (LQTS2) und SCN5A (LQT3).
Jervell- und Lange-Nielsen-Syndrom
Es ist die autosomal-rezessive Form von LQTS.
Es ist mit angeborener neurologischer Taubheit oder anderen angeborenen Herzerkrankungen, Autismus, vollständiger Syndaktylie und Immunschwäche verbunden.
Es wird speziell durch eine Mutation in den Genen KCNE1 und KCNQ1 verursacht.
Unbehandelt sterben etwa 50 % im Alter von 15 Jahren an ventrikulären Arrhythmien.
Romano-Ward-Syndrom
Das Romano-Ward-Syndrom ist die autosomal dominante Form des LQTS.
Es ist nicht mit angeborener neurologischer Taubheit oder anderen angeborenen Herzerkrankungen, Autismus, vollständiger Syndaktylie und Immunschwäche verbunden.
Diagnose und Werte des Long-QT-Syndroms:
Die Diagnose von LQTS ist oft nicht einfach, da 2.5 % der gesunden Bevölkerung ein verlängertes QT haben und 10-15 % der LQTS-Patienten ein normales QT-Intervall haben: oft können sogar einige Profisportler es haben, ohne dass medizinisches Personal es bemerkt.
Ein häufig verwendetes diagnostisches Kriterium basiert auf dem LQTS „diagnostic score“.

Die Punktzahl wird durch die Vergabe von Punkten nach verschiedenen unten aufgeführten Kriterien berechnet.
Bei 4 oder mehr Punkten ist die Wahrscheinlichkeit für LQTS hoch, bei 1 Punkt oder weniger ist die Wahrscheinlichkeit gering; 2 oder 3 Punkte zeigen eine mittlere Wahrscheinlichkeit an.
QTc (definiert als QT-Intervall/Quadratwurzel des RR-Intervalls)
- >= 480 ms – 3 Punkte
- 460-470 ms – 2 Punkte
- 450 ms und männliches Geschlecht – 1 Punkt
- Ventrikuläre Tachykardien vom Typ Torsades de Pointes – 2 Punkte
- T-Wellenwechsel – 1 Punkt
- T-Wellen-Lawine in mindestens 3 Ableitungen im EKG – 1 Punkt
- Niedrige Herzfrequenz für das Alter (Kinder) – 0.5 Punkte
- Synkope (für Synkope und Peak Torsion im selben Fach können keine Punkte vergeben werden)
- Unter Stressbedingungen – 2 Punkte
- Außerhalb von Stressbedingungen – 1 Punkt
- Angeborene Taubheit – 0.5 Punkte
- Familienanamnese (dasselbe Familienmitglied kann nicht sowohl für plötzlichen Tod als auch für LQTS gezählt werden)
- Andere Familienmitglieder mit eindeutiger Diagnose von LQTS – 1 Punkt
- Plötzlicher Tod naher Familienangehöriger (Mitglieder unter 30 Jahren) – 0.5 Punkte
Therapie
Bei asymptomatischen Patienten ohne Nachweis ventrikulärer Arrhythmien und ohne positive Familienanamnese für plötzlichen Tod wird eine alleinige Beobachtung und möglicherweise eine medikamentöse Therapie empfohlen, ohne dass die maximal verträgliche Dosis erreicht werden muss.
Bei LQTS1 und LQTS2 können Betablocker verwendet werden; bei LQTS 3 sind Klasse-Ib-Antiarrhythmika wie Mexiletin vorzuziehen.
Die gleichen Medikamente können zur Behandlung von Notfallpatienten verwendet werden, mit der Einschränkung, dass nur Lidocain verwendet werden sollte, bis die Diagnose von LQTS1 oder LQTS2 bestätigt ist, da Betablocker Arrhythmien bei LQTS3-Patienten verschlimmern können.
Eine antiarrhythmische Therapie sollte jedoch bei Patienten, die asymptomatisch sind, aber Anzeichen für nicht anhaltende ventrikuläre Arrhythmien und eine Familienanamnese mit plötzlichem Tod aufweisen, in der maximal tolerierten Dosis angewendet und durchgeführt werden.
Implantierbarer Kardioverter Defibrillator (ICD)-Implantation wird bei diesen Patienten nicht unbedingt empfohlen.
Letzteres ist jedoch unbedingt indiziert (Klasse-I-Indikation) bei Patienten mit Synkopen oder bereits durchlebten Herzstillständen.
Die Schrittmacherfunktion des Geräts muss bei Patienten ausgenutzt werden, die Arrhythmien während Bradykardie oder Herzrhythmuspausen aufweisen.
Bei Patienten, die trotz optimaler medikamentöser Therapie noch Symptome aufweisen, ist eine links-zervikal-thorakale Gangliektomie mit Zerstörung des Ganglion stellatum und der ersten drei oder vier thorakalen sympathischen Ganglien indiziert.
Prognose und Risiko
Bei unbehandelten LQTS-Patienten kann das Risiko eines Ereignisses (Synkope oder Herzstillstand) anhand des Genotyps (LQTS1-10), des Geschlechts und des QTc-Intervalls abgeschätzt werden.
Hohes Risiko (>50%)
QTc > 500 ms LQTS1 & LQTS2 & LQTS3 (Männer)
Mittleres Risiko (30-50%)
QTc > 500 ms LQTS3 (Frauen)
QTc < 500 ms LQTS2 (Frauen) & LQTS3
Geringes Risiko (<30%)
QTc < 500 ms LQT1 & LQT2 (Männer)
Long-QT-Syndrom und Sport
Patienten mit LQTS können unter strenger kardiologischer Kontrolle weiterhin Sport treiben, vermeiden jedoch Sportarten mit längerer hoher Anstrengung und Wassersportarten wie Schwimmen und Tauchen.
Lesen Sie auch:
Herzkrankheit: Was ist Kardiomyopathie?
Entzündungen des Herzens: Myokarditis, infektiöse Endokarditis und Perikarditis
Herzgeräusche: Was es ist und wann man sich Sorgen machen sollte
Broken-Heart-Syndrom ist auf dem Vormarsch: Wir kennen die Takotsubo-Kardiomyopathie
Was ist ein Kardioverter? Überblick über den implantierbaren Defibrillator
Erste Hilfe bei Überdosierung: Notarzt rufen, was tun, während auf die Retter gewartet wird?
Squicciarini Rescue wählt Emergency Expo: American Heart Association BLSD- und PBLSD-Schulungskurse
'D' für Deads, 'C' für Kardioversion! – Defibrillation und Fibrillation bei pädiatrischen Patienten
Herzentzündungen: Was sind die Ursachen einer Perikarditis?
Thrombose kennen, um in das Blutgerinnsel einzugreifen
Patientenverfahren: Was ist externe elektrische Kardioversion?
Erhöhung der Belegschaft von EMS, Schulung von Laien in der Verwendung von AED
Unterschied zwischen spontaner, elektrischer und pharmakologischer Kardioversion
Was ist Takotsubo-Kardiomyopathie (Broken-Heart-Syndrom)?
Das EKG des Patienten: So lesen Sie ein Elektrokardiogramm auf einfache Weise ab
Belastungstest, der bei Personen mit LQT-Intervall ventrikuläre Arrhythmien induziert