
Family hypercholesterolemia: what it is and how to treat it
Familial hypercholesterolemia, a rare genetic disease that causes serious health consequences (e.g. heart attack and stroke) that require appropriate containment measures with appropriate medication and an appropriate lifestyle.
What does hypercholesterolemia mean? And what does familial mean?
Hypercholesterolemia is a clinical condition characterized by excessive cholesterol in the blood.
Cholesterol is a component of lipids, usually taken with the diet but which can also be produced by the body.
Cholesterol is very important for life because it is used for the formation of cell membranes (to ensure their function), protects neurons and cranial nerves.
Cholesterol is also used for the synthesis of other molecules, three of which are bile acids (important for digestion), some hormones, and Vitamin D.
Only if present above these requirements can it cause serious damage.
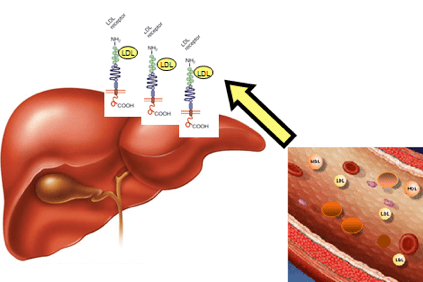
In the case of hypercholesterolemia, cholesterol accumulates in the blood in the form of light lipoproteins (LDL), particular aggregates of fats and proteins, also called “bad cholesterol” that promote the formation of plaques in the artery wall (atherosclerotic plaques).
This happens when patients have such high cholesterol levels that they cannot eliminate it through the physiological mechanisms of the liver.
The accumulation of LDL in the blood eventually leads to the formation of the atheroma (a physical obstruction to normal blood flow) which can lead to more serious consequences such as angina, heart attack, stroke, but also in other organs such as the brain, kidneys, lungs and the liver itself.
Hypercholesterolemia is familiar when transmitted to offspring
This is due to modifications of a gene that contains the information to make a liver protein, the LDLR (LDLR) receptor, which recognizes LDL, removes it from the bloodstream, and transports it to the liver cells, which then remove it. This gene alteration causes LDL accumulation in the blood.
To date, there are more than 600 known alterations in the LDL gene that cause family hypercholesterolemia.
Also, people with this disorder can bring changes in other genes involved in lipoprotein metabolisms such as the APOB gene (apoprotein B in LDL) and the PCSK9 gene (a protein that degrades LDL receptors).
Although it is known that cholesterol accumulation can be harmful, it is estimated that up to one-third of heart attacks occurring before age 40 are due to undiagnosed or inappropriately treated family hypercholesterolemia.
More common is multifactorial hypercholesterolemia caused by environmental factors (a diet high in fat, especially if associated with physical inactivity) even in the presence of predisposing genetic factors.
The forms which family hypercholesterolemia manifests itself
This disease can present itself in 2 different forms: a less serious one (heterozygous, 1 case every 500 individuals) and a more serious one (homozygous, 1 case every 1,000,000 individuals)1,2.
The heterozygous form is often asymptomatic and is diagnosed only based on blood cholesterol levels.
The liver struggles to eliminate LDL because LDL receptors are produced in insufficient numbers, resulting in a 2 or 3-fold increase in blood levels compared to normal values.
This form can lead to an increased risk of cardiovascular disease in adulthood.
The homozygous form is characterized by the onset of cardiovascular disease even at a young age and the presence of characteristic accumulations of fat such as xanthomas (yellowish nodules on the knuckles of the hands and Achilles tendon) and xanthelasma (yellowish plates on the eyelids and around the eyes).
The genetic defect is inherited from both parents and, the risk of a heart attack in the absence of therapy is already observed around 15-20 years of age.
In fact, in this condition, the liver fails to metabolize the lipoproteins that remain in the blood, which then accumulate leading to the dysfunctions described above and create a situation incompatible with life.
What are the chances of transmission of the disease to children?
Since each of us has two copies of each gene, each inherited from a parent, we will have the heterozygous form when we inherit one altered and one healthy copy.
On the contrary, we will have the homozygous form if we inherit both copies of the diseased gene from both parents. It must be said that this form is very rare because it occurs only when both parents have the gene that causes the disease and therefore each of them can transmit a copy of the same gene alteration.
Familial hypercholesterolemia is called heterozygous pathology composed in the rare cases in which two different types of gene alterations are inherited, one from each parent.
Children at risk of developing the disease must be diagnosed and treated early. Today it is possible to diagnose the disease through genetic tests that look for errors (gene alterations) on LDLR, ApoB, and PCSK9 genes.
Therefore only the forms of family hypercholesterolemia are transmissible.
However, multifactorial forms that occur mainly due to an incorrect lifestyle (accumulation of cholesterol with the diet), can also be characterized by a genetic predisposition.
In fact, these subjects may be present genetic deficits that compromise the body’s ability to adequately compensate for excess lipid in the diet.
For example, when the liver is saturated with cholesterol taken with food, the production of receptors that capture LDL circulating in the blood may be suppressed.
A situation is very similar to the one that occurs during family hypercholesterolemia although less severe.
In this case, the concentration of total cholesterol in the blood is above normal and is usually between 240 and 350 mg/dl compared to the normal one conventionally set around 200 – 240 mg/dl.

A curiosity: this disease may have been handed down over the centuries
A careful observation of the famous painting of the Mona Lisa shows the possibility that Mona Lisa already at a young age suffered from family hypercholesterolemia.
In fact, Leonardo also faithfully depicts the typical fat deposits on the hands and near the eyes (now known as xanthomas and xanthelasma) that indicate the presence of the pathology with certainty.
If this is true, it is shown that family hypercholesterolemia is a disease preserved over the centuries (a case documented as early as 1500).
What are the possibilities to treat this disease today?
The possibility of curing the disease depends on its severity. Also, risk factors (diet, smoking, age, family, and personal history of hypercholesterolemia, presence of other diseases) can worsen the overall disease picture.
To treat family hypercholesterolemia, new-generation drugs can be used, but at the same time, it is necessary to act with an appropriate lifestyle modification.
In cases of intermediate severity (heterozygous family hypercholesterolemia), statin-based drug therapy (cholesterol synthesis inhibitors that induce increased LDLR receptor synthesis) and the combination with ezetimibe (cholesterol absorption inhibitors) or PCSK9 inhibitory drugs (i.e. PCSK9 protein inhibitors whose role is to destroy liver receptors that capture LDLR) improves the activity of the healthy copy of the LDLR gene and reduces the accumulation of cholesterol in the blood.
Recently approved is also bempedoic acid, a drug that acts in the liver by inhibiting the enzyme ATP citrate lyase, a molecule involved in the process of endogenous cholesterol synthesis.
This mechanism of action allows us to act on the amount of cholesterol produced, acting upstream from the site of action of the statins, and to stimulate the expression of LDL receptors to compensate for the reduced synthesis.
Unlike statins, bempedoic acid is not active in skeletal muscle, which decreases the possibility of having unwanted events typical of statins.
This drug can also be associated with ezetimibe with favorable effects.
Homozygous familial hypercholesterolemia was until recently considered an incurable disease.
Statin-based therapy is not effective in this pathology. In fact, statins that act on the mechanisms that lead to the production of endogenous cholesterol cannot stimulate the synthesis of LDL receptors.
To eliminate LDL cholesterol from the body of these patients, plasmapheresis, a technique that allows filtering the blood by eliminating fats, similarly to what is done with dialysis when the kidneys do not work.
However, this is an invasive procedure with a negative impact on patients’ quality of life.
The latest research on family hypercholesterolemia
In recent years, research has led to the development of specific drugs also for the homozygous form of family hypercholesterolemia, greatly improving the expectation and quality of life of patients who suffer from it.
For example, the drug lomitapide (taken orally) leads to markedly lower plasma LDL-cholesterol levels in these patients.
Lomitapide inhibits a microsomal triglyceride transport protein (MTP) that allows the incorporation of these lipids together with the apoprotein B100 in nascent VLDLs.
As a result, the apoprotein B100 and LDL are reduced even in patients where the disease has caused the total absence of the LDL receptor.
Another new generation drug is mipomersen, an antisense oligonucleotide capable of degrading the B100 apoproteins that take part in the formation of LDL, thus reducing the number of the latter.
Evolocumab and alirocumab are two monoclonal antibodies that inhibit the activity of the PCSK9 protein in the blood.
However, for these two drugs to take effect, at least a small part of the LDL receptors must be present and functioning.
Among the drugs still in clinical development is the inclusion of a biological drug (so-called siRNA) that blocks DNA transcription and synthesis of the PCSK9 protein in the liver.
Positive results have been recently achieved with evinacumab, a monoclonal antibody inhibitor of the protein similar-angiopoietin 3 (ANGPTL3), a molecule synthesized by the liver whose role is to increase cholesterol-LLDL and triglyceride levels preventing their degradation.
This new drug has also shown efficacy in patients where the LDL receptor is not functioning3,4.
Recently some attempts has also been made for understanding if gene therapy, with the introduction of the healthy gene into the patient’s DNA, could work5.
From this list of possible therapies for familial hypercholesterolemia, it is possible to understand how much research is being done to provide solutions for these patients who, although rare, are afflicted by this serious disease, reducing the risk of fatal consequences.
However, as always happens with new drugs, it is good to use a word of caution on their chronic use since the knowledge about their possible side effects will be clearer only with experience (pharmacovigilance), so their use in pediatrics should be carefully evaluated, of course with the ultimate aim of offering these “little” patients the possibility to lead a “normal” life.
Bibliographic and sitographic references for the article on family hypercholesterolemia
1 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Authors/Task Force Members; ESC Committee for Practice Guidelines (CPG); ESC National Cardiac Societies. Atherosclerosis. 2019 Nov;290:140-205. doi: 10.1016/j.atherosclerosis.2019.08.014.
2 Rare dyslipidaemias, from phenotype to genotype to management: a European Atherosclerosis Society task force consensus statement. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG, Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet Diabetes Endocrinol. 2020 Jan;8(1):50-67. doi: 10.1016/S2213-8587(19)30264-5.
3 Lipoprotein(a) Lowering-From Lipoprotein Apheresis to Antisense Oligonucleotide Approach. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica M.J Clin Med. 2020 Jul 3;9(7):2103. doi: 10.3390/jcm9072103.
4 LDL-Cholesterol-Lowering Therapy. Pirillo A, Norata GD, Catapano AL.Handb Exp Pharmacol. 2020 Apr 30. doi: 10.1007/164_2020_361.
5 A review of gene- and cell-based therapies for familial hypercholesterolemia. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A.Pharmacol Res. 2019 May;143:119-132. doi: 10.1016/j.phrs.2019.03.016.