
Scleroderma, how it manifests itself and what treatments are available
Scleroderma is a rare autoimmune disease (10-20 cases per 1,000,000 people), localised mainly in the skin, with very different symptoms from one patient to another
It begins between the ages of 30 and 50, although it does not spare other age groups, and there is a clear prevalence in the female sex (4:1).
There is a wide range of clinical expression.
A classification, based on extension and severity, distinguishes between localised forms (20 times more frequent) and more severe, diffuse forms in which internal organs are also involved (systemic scleroderma).
What is the cause of scleroderma?
There is no known underlying cause.
In this disease, the immune system attacks the endothelial cells (which line the inside of blood vessels) and stimulates an uncontrolled production of collagen, a component of the connective tissue that provides support and nourishment for all organs.
Collagen is the substance found in scars. Its uncontrolled accumulation creates a ‘hardening’ of areas of the skin, i.e. fibrosis (hence the term sclero-dermia = hard skin); when it involves (in extensive forms) other organs of the body, it causes progressive damage to their function.
Scleroderma is, of course, not contagious.
What are the symptoms of scleroderma?
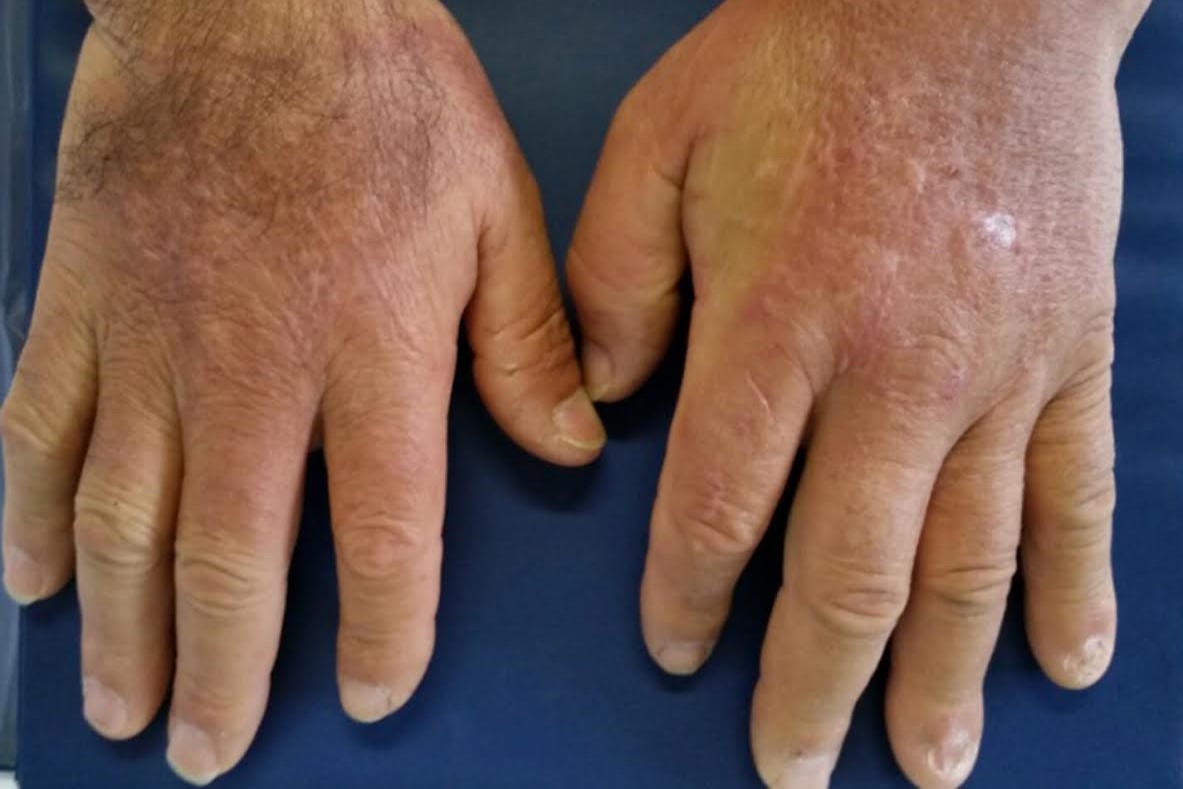
In localised forms (also called Morphea) it begins with red-purple patches that tend to progressively harden; the central part of these spots becomes lighter, almost whitish, surrounded by a pinkish border; the final appearance (even after years) is that of a scar, darker or lighter (hyper- or hypopigmented).
Several variants exist:
- scalp morphea (with light scars and total disappearance of hair or alopecia);
- a form known as ‘drop-shaped’, with an appearance of small, roundish spots;
- a linear form, which tends to deepen in the subcutaneous tissue, down to the muscle and tendons; they occur more in the arms and legs, but do not spare the face (‘sabre-like’ appearance)
- the annular form (rarer), localised more frequently to the genitals;
- the deep morphea, which is more aggressive and rapidly involves the skin, muscles and bones.
The symptoms of Systemic Scleroderma are very different from Morphea and the severity varies from case to case
Two types can be distinguished:
A) Limited Systemic Scleroderma, in which the main and initial disorder is Raynaud’s phenomenon.
This is a circulatory disorder that mainly affects the fingers of the hands, triggered mainly by cold, and is manifested by alternating pallor (due to vasospasm), cyanosis and then erythema; with time, chronic damage of the fingers is created, which may extend to the forearms and face.
In 25 % of these patients, five complications known as CREST occur:
- Subcutaneous calcifications;
- Raynaud’s (also in the ears, nose and other skin areas);
- Oesophagitis (damage to the digestive area that is between the mouth and the stomach), with difficulty in passing food;
- Sclerodactyly (thickening of the skin of the fingers, with enlargement, creating a ‘sausage’ deformity; sometimes affecting the neck, ears and face;
- Telangiectasias (dilation of small blood vessels located on the skin of the hands, face and lips).
CREST is a disease with a very slow evolution and does not extend to internal organs.
B) Diffuse systemic scleroderma: this form begins with Raynaud’s phenomenon but rapidly (after a few months) extends to the internal organs.
Raynaud’s phenomenon is much more severe, with ulcerations of the skin, also extending to the legs, joints, etc.
The organs most affected are the digestive system (oesophagus, intestines and rectum, with difficulty in swallowing, malabsorption and severe difficulty in evacuation) bones and joints (with painful arthritis), muscles (cramps, muscle pain and tendonitis), kidneys (up to kidney failure), heart and lungs.
Diffuse systemic scleroderma is a very serious and rapidly progressive disease and must be recognised early.
How to recognise the first symptoms of the disease?
Morphea can be recognised by the appearance of areas of hardening of the skin, of different shapes, with a very slow progression.
The systemic forms (both localised and diffuse) in 95% of cases begin with Raynaud’s phenomenon: sudden changes in the superficial circulation of the fingers (alternating pallor and bluish or cyanotic appearance and redness) should be an alarm bell.
Tingling and pain may be present.
The phenomenon (also present in other connective tissue diseases) is related to blood vessel damage, with an initial vasospasm, mainly due to a drop in external temperature (pallor).
This is followed by cyanosis, due to reflex vasodilation, and finally a more prolonged reddening.
In systemic cases, there may be

- swallowing and digestive difficulties (up to actual malabsorption);
- the appearance of calcifications and increased consistency of the skin;
- joint or muscle pain;
- hypertension (due to kidney damage);
- difficulty breathing (pulmonary, pleural but also cardiac involvement).
What are the examinations and tests for the diagnosis of scleroderma?
In the case of circulatory disorders, capillaroscopy is useful, on prescription from the dermatologist/rheumatologist specialist: this is a microscopic examination of the capillaries of the nail matrix that allows their alterations to be studied.
In systemic scleroderma, the presence of mega-capillaries and areas where blood vessels are absent (avascular areas) is found.
Antinuclear antibodies (especially anti-centromere) and, in diffuse systemic forms, anti-Scl70 antibodies can be sought in laboratory tests.
Analyses do not always help recognise the early stages of scleroderma and therefore a specialist visit (Rheumatologist and Dermatologist) is essential to recognise early symptoms and assess possible involvement of the various organs.
How can scleroderma be prevented?
There is no real prevention, nor are there any lifestyle habits that can reduce the risk of contracting the disease or aggravate it.
The only prevention consists in recognising the early symptoms (starting with Raynaud’s phenomenon) and resorting to a thorough specialist examination.
In cutaneous forms it is necessary to reduce exposure to the sun or use high photoprotection, as this causes aggravation.
How is scleroderma treated?
Cutaneous forms (morphea) progress very slowly and not severely, being limited to the skin.
The outcome is mainly ‘unsightly’ because the disease’s effects are permanent.
Products for local use are mainly used to slow down/stop the course of the disease in the affected areas, mainly cortisone, moisturising and emollient creams.
Phototherapy is also used.
The annular form (localised mainly in the penis) may lead to a circulatory problem and in some cases require surgical treatment if local therapy has not been beneficial.
In systemic scleroderma, immunosuppressive drugs are used; in recent years, in addition to cortisone drugs, the use of biological drugs has increased, with promising results on the overall course of the disease.
There are also specific treatments for the internal organs attacked by the disease (heart, oesophagus and digestive system, lungs, kidneys, etc.), to limit the damage of the disease and alleviate the symptoms.
Read Also:
Emergency Live Even More…Live: Download The New Free App Of Your Newspaper For IOS And Android
Psoriasis, An Ageless Skin Disease
Relapsing-Remitting Multiple Sclerosis (RRMS) In Children, EU Approves Teriflunomide
ALS: New Genes Responsible For Amyotrophic Lateral Sclerosis Identified
Psoriatic Arthritis: What Is It?
Rehabilitation Therapies In The Treatment Of Systemic Sclerosis
Psoriasis: It Gets Worse In Winter, But It’s Not Just The Cold That’s To Blame
Exposure To Cold And Symptoms Of Raynaud’s Syndrome
Scleroderma. Blue Hands, A Wake-Up Call: The Importance Of Early Diagnosis
Scleroderma: Causes, Symptoms And Treatment