
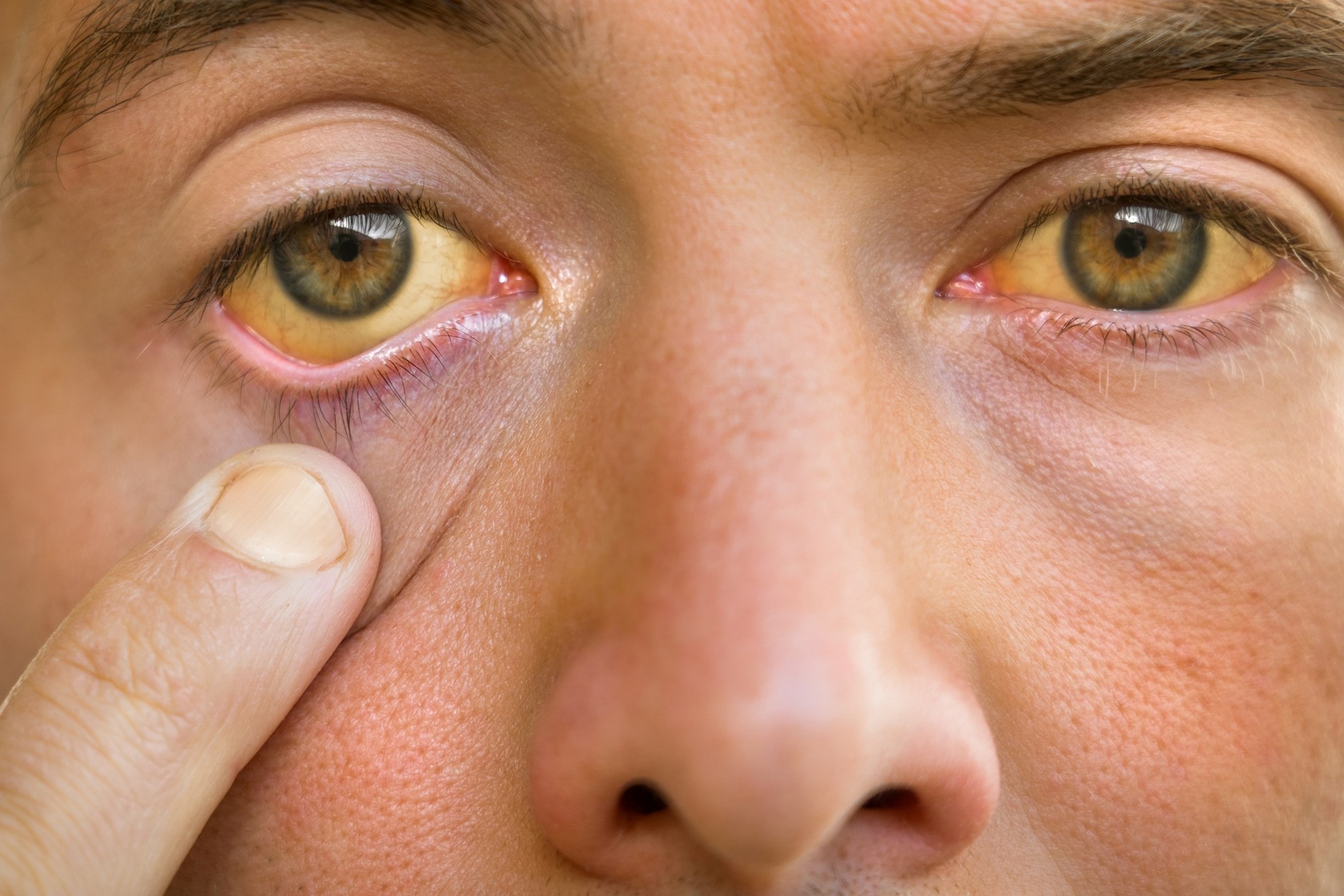
What is Gilbert's Syndrome
Gilbert’s syndrome is a familial and hereditary pathophysiological condition, of little clinical relevance and with a benign prognosis, characterised by jaundice or hepatic sub-artery, caused by a congenital alteration of bilirubin metabolism and consequent increase in bilirubinemia in the blood, i.e. the presence of hyperbilirubinemia (with scleral jaundice or sub-artery)
Hyperbilirubinemias manifest themselves with jaundice and are differentiated according to various different types.
Jaundice is classified into three species: pre-hepatic, hepatic and post-hepatic.
Gilbert’s syndrome is a jaundice (or sub-atherectomy if less manifest) of the hepatic type, with varying intensity
It depends on an altered function within the hepatocyte, the main cell of the liver; it is a congenital and familial enzymatic defect, which leads to a dysfunction and reduction of the normal process of glucurono-conjugation of bilirubin, resulting in an excess of indirect bilirubin in the serum and in the composition of bile.
Bilirubin is the end product of normal heme degradation, it comes from the catabolism of heme contained in the senescent red blood cell (each red blood cell lasts about 100 days, in Gilbert’s even less); bilirubin production takes place in the reticuloendothelial system, mainly spleen and bone marrow, after which it is taken up by hepatocytes in the liver that subject it to the glucurono-conjugation process, which is defective in Gilbert’s syndrome.
Thus, bilirunin is released in an indirect or ‘unconjugated’ form.
This disorder of bilirubin metabolism can have varying degrees of severity, from mild as in Gilbert syndrome, to severe as in the elevated hyperbilirubinemia of Crigler-Najjar I and II syndromes, fortunately less frequent and rare.
How Gilbert’s syndrome manifests itself
Gilbert’s syndrome is a fairly frequent condition (approximately 5 – 8% of the Italian population).
Sufferers present with a usually fluctuating increase in serum bilirubinemia, especially indirect or unconjugated bilirubinemia (sometimes also direct bilirubinemia but, in this case, not prevalent).
The rate of bilirubinemia, and of unconjugated bilirubin, is generally highly variable, and is often within the normal range in many individuals with Gilbert syndrome.
It is a hereditary alteration that does not create any type of problem on either the quality or the length of life of sufferers.
Under certain conditions (fever, infections, intercurrent, stress, alcohol abuse, physical exertion, fasting) there can be a marked increase, even above the value of 2.5 – 5 mg/100 ml of bilirubinemia, which, however, promptly returns to normal when these particular, usually temporary, situations cease.
Diagnosis of Gilbert’s syndrome
Gilbert syndrome is discovered in the subject mostly by chance, mostly during laboratory investigations performed for routine or occasional checks.
The age of onset of a first jaundice or subictal episode is usually around 15-18 years.
In 40% of individuals with Gilbert syndrome, there is a slight decrease in red blood cell lifespan, which may lead to a suspicion of haemolysis.
The diagnosis of Gilbert’s syndrome first requires the exclusion of any other hepatocellular, biliary, haemolytic disease that may cause (or concause) jaundice; in addition, an increase in bilirubinemia, predominantly indirect bilirubinemia at least three times in succession, two or three findings of increased serum values at intervals of a few months, concomitant with absolute normality of all liver function tests (such as transaminases, gGT, alkaline phosphatase, direct and indirect bilirubinemia, bile acids, blood count) is required, i.e. the presence of isolated hyperbilirubinemia.
A normal abdominal ultrasound may also be useful and necessary to ascertain the normality of the liver and the intra- and extra-hepatic biliary tracts.
It is very difficult for a liver biopsy to be performed for such a diagnosis, except perhaps in the case of seeking a more thorough differential diagnosis in problematic or intricate situations.
The differential diagnosis, in order to distinguish Gilbert syndrome from other pathologies with some common features, must be made above all with the other hereditary pathologies affecting bilirubin metabolism, namely, as mentioned earlier, Crigler-Najjar I syndrome, which is much more serious and has an inauspicious prognosis; Crigler-Najar II syndrome, less worrying and with a relatively good prognosis but with higher indirect hyperbilirubinemia values than Gilbert’s and earlier onset, mostly in childhood, unlike Gilbert’s, in which the onset of jaundice usually appears later, i.e. around 15-18 years of age.
Rotor syndrome and Dubin-Johnson syndrome are also familial and hereditary hyperbilirubinemias but with increased direct or conjugated bilirubin
They are also pathologies of bilirubin metabolism, very rare conditions, but with different pathogenetic mechanisms, also with more intense jaundice; occasionally they can also be found in pregnancy or during oestrogen therapy, sometimes to be distinguished from post-hepatic-type jaundice, but with normal cholestasis indices.
It is worth mentioning other forms of morbid conditions characterised by increased unconjugated (or indirect) bilirubinemia in the blood as in Gilbert’s syndrome: physiological jaundice of the newborn and breast milk jaundice, which occur at birth and are benign and momentary in nature. There is, respectively, a delayed maturation of bilirubin metabolism and, in the second case, the presence of prolongation of physiological jaundice due to a temporary alteration of glycuronoconjugation by predisposition and with genetic alterations similar to those of Gilbert’s syndrome.
Other conditions of altered bilirubin metabolism include hyperbilirubinemia by drugs (e.g. sulphonamide – a bacteriostatic – or fusidic acid – an antibiotic).

Recently, several forms of liver damage have been described in patients with familial hyperbilirubinukaemia, particularly in Gilbert; these patients should be closely monitored from the point of view of liver function when they require treatment with numerous drugs, or in the case of chemotherapy.
Finally, for the sake of completeness, with regard above all to jaundice, the whole large chapter of acute and chronic hepatopathies (hepatitis), obstructions of the biliary tree, and the whole chapter of cholestasis, with regard to bile, biliary secretion, conditions of hepatocyte involvement, and lastly the large chapter of haemolysis, in which hyperbilirubinemia is found together with alterations in the various haematochemical haematological parameters.
Prognosis of Gilbert syndrome
The prognosis of Gilbert’s syndrome is, as mentioned above, good and even excellent.
There are, in fact, no adverse effects on liver function or on the well-being of the entire organism.
Therapy for Gilbert’s syndrome
No therapy is necessary.
As a measure to avoid any temporary increase in hyperbilirubinaemia, it is recommended to
- reduce or prevent stress
- avoid fasting
- do not abuse alcohol
- avoid excessive physical exertion.
Read Also:
Emergency Live Even More…Live: Download The New Free App Of Your Newspaper For IOS And Android
Hepatitis In Children, Here Is What The Italian National Institute Of Health Says
Gilbert’s Syndrome: Symptoms, Causes And Diagnosis Of This Liver Disease
Acute Hepatitis In Children, Maggiore (Bambino Gesù): ‘Jaundice A Wake-Up Call’
Nobel Prize For Medicine To Scientists Who Discovered Hepatitis C Virus
Hepatic Steatosis: What It Is And How To Prevent It
Acute Hepatitis And Kidney Injury Due To Energy Drink Consuption: Case Report
The Different Types Of Hepatitis: Prevention And Treatment
Acute Hepatitis And Kidney Injury Due To Energy Drink Consuption: Case Report
New York, Mount Sinai Researchers Publish Study On Liver Disease In World Trade Center Rescuers
Acute Hepatitis Cases In Children: Learning About Viral Hepatitis
Hepatic Steatosis: Causes And Treatment Of Fatty Liver
Hepatopathy: Non-Invasive Tests To Assess Liver Disease
Liver: What Is Non-Alcoholic Steatohepatitis