
Malattie rare: la Sindrome di Rothmund-Thomson
La sindrome di Rothmund-Thomson è una malattia della pelle molto rara che sin dalla sua prima descrizione in Austria nel 1868, è stata descritta in circa 500 individui
Manifestazioni della sindrome di Rothmund-Thomson
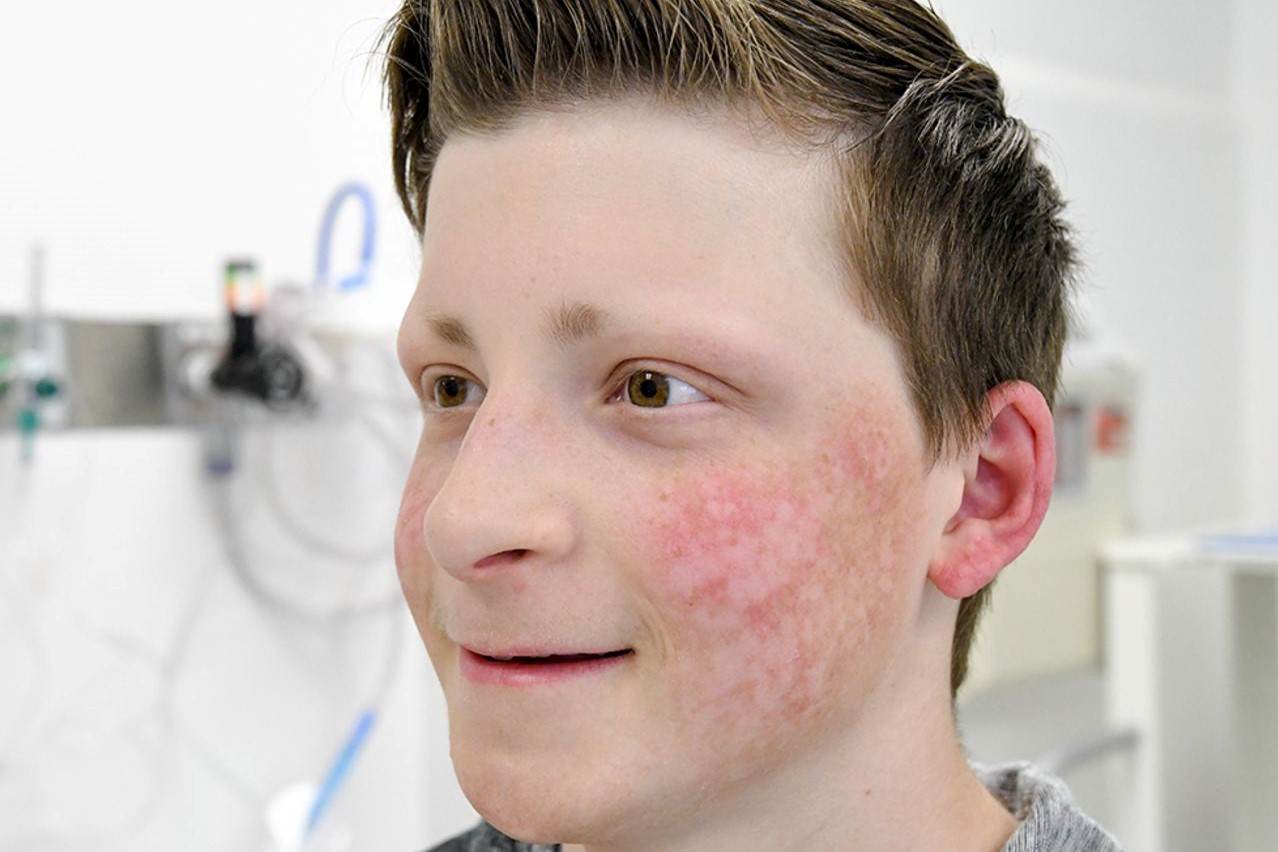
Si manifesta con il poichiloderma, una tipica eruzione cutanea del viso caratterizzata da un assottigliamento (atrofia) dello strato più esterno della pelle (l’epidermide), anomalie della colorazione (pigmentazione) della pelle sia con ipo che iperpigmentazione e dilatazione dei vasi superficiali della pelle (teleangectasie).
Oltre all’interessamento cutaneo, sono presenti sintomi extracutanei quali un ritardo di crescita sia pre che postnatale, bassa statura, capelli ciglia e/o sopracciglia rade o assenti, anomalie dei denti e scheletriche, cataratta giovanile, invecchiamento precoce e predisposizione al cancro (soprattutto osteosarcoma).
La sindrome di Rothmund-Thomson è una malattia causata nella maggior parte dei casi da alterazioni (mutazioni) del gene dell’elicasi RECQL4 che si trova sul cromosoma 8
Si trasmette come carattere autosomico recessivo.
Le malattie a trasmissione autosomica recessiva si verificano soltanto nelle persone che hanno ereditato due copie alterate (mutate) di un gene.
Sono mutate sia la copia ereditata dalla mamma sia la copia ereditata dal papà.
Il termine “recessivo” significa che l’alterazione di una sola delle due copie di geni non è sufficiente per provocare la malattia.
Per causare la malattia, entrambe le copie dei geni debbono essere mutate.
I genitori sono portatori di una copia sola del gene alterato (l’altra copia è normale) quindi non sono malati: sono portatori sani.
Due portatori sani che vogliono avere figli hanno, a ogni gravidanza, il 25% di probabilità (una su quattro) di avere un figlio malato.
Il 50% dei loro figli sarà portatore sano (come la madre e il padre, senza sintomi) e il restante 25% sarà sano (con entrambe le copie del gene senza la mutazione).
Queste probabilità sono indipendenti dal sesso del nascituro.
Ad oggi, mutazioni nel gene RECQL4 sono state identificate nel 60-65% circa dei pazienti, mentre nel restante 35-40% degli individui affetti la causa non è nota.
I segni clinici che possono presentare i pazienti affetti dalla sindrome di Rhotmund-Tomson includono una eruzione cutanea
Di solito quest’ultima non è presente alla nascita ma si sviluppa tra i 3 e i 6 mesi di vita con un arrossamento della pelle (eritema), rigonfiamento e cicatrizzazioni sul viso e successiva diffusione dell’eruzione ai glutei e alle estremità, mentre in genere il tronco e l’addome non vengono colpiti.
L’eruzione cutanea evolve dopo anni nel tipico poichiloderma con aree di iperpigmentazione alternate ad aree di ipopigmentazione, piccole chiazze di assottigliamento (atrofia) e dilatazione dei vasi supeficiali (teleangiectasie).
Possono essere presenti anomalie scheletriche degli arti superiori con assenza o malformazioni del pollice o con un accorciamento degli avambrambracci.
Altre frequenti anomalie scheletriche sono l’assenza (aplasia) o una abnorme riduzione delle dimensioni (ipoplasia) della rotula e l’osteopenia, vale a dire assottigliamento e indebolimento delle ossa.
Possono essere presenti anche una bassa statura e un basso peso corporeo, anomalie dentali, disturbi gastrointestinali, displasia delle unghie, cataratta bilaterale, tumori tra cui l’osteosarcoma.
La diagnosi di sindrome di Rothmund-Thomson si formula sulla base di un’attenta raccolta della storia clinica e su un altrettanto attenta visita del bambino.
A indirizzare verso la diagnosi è soprattutto la presenza del poichiloderma
La diagnosi va presa in considerazione anche nei pazienti affetti da osteosarcoma associato ad alterazioni della pelle.
Nei casi in cui l’eruzione cutanea risulti atipica, la diagnosi clinica può essere formulata quando sono presenti almeno altre due caratteristiche della sindrome di Rothmund-Thomson tra cui:
- Capelli, sopracciglia e/o ciglia sparse e rade;
- Bassa statura proporzionata a un basso peso;
- Disturbi gastrointestinali presenti già dall’età infantile, quali vomito cronico e diarrea;
- Anomalie scheletriche quali difetti del radio, dell’ulna, assenza o ipoplasia della rotula, osteopenia;
- Anomalie dei denti (denti poco sviluppati, difetti dello smalto o ritardo nella dentizione);
- Sviluppo anomalo delle unghie (unghie displasiche);
- Ispessimento (ipercheratosi) della pianta dei piedi;
- Cataratta, di solito giovanile, bilaterale;
- Tumori, inclusi tumori della pelle (carcinoma baso-cellulare e a cellule squamose) e l’osteosarcoma.
La diagnosi viene poi confermata con l’analisi molecolare del gene RECQL4 (sequenziamento dell’intero gene).
Quando sono presenti i criteri clinici che consentono di diagnosticare la sindrome di Rothmund-Thomson, le mutazioni del gene RECQL4 vengono identificate solo nel 60-65% dei casi.

Nel rimanente 35-40% dei casi non è stato ancora possibile identificare un altro gene responsabile.
Non esiste un trattamento risolutivo.
I bambini affetti dalla sindrome di Rothmund-Thomson devono essere seguiti sin dalla nascita da un’équipe multidisciplinare che includa anzitutto la presenza di specialisti per la gestione delle problematiche della cute e l’utilizzo di un trattamento laser delle lesioni teleangectasiche.
Dell’équipe deve anche far parte un oculista per effettuare visite oculistiche annuali (per valutare la presenza della cataratta e per la sua eventuale correzione chirurgica).
Indagini di imaging come radiografie e TC sono indispensabili in caso di sintomi potenzialmente associati a tumore osseo.
Un supplemento con calcio e vitamina D può essere considerato nei pazienti con osteopenia e/o fratture.
I pazienti devono evitare l’esposizione al calore e al sole che potrebbe aggravare l’eruzione cutanea in alcuni individui e aumentare il rischio di sviluppare un tumore alla pelle.
Dato il teorico rischio di sviluppare tumori, l’ormone della crescita (GH) non è raccomandato per i soggetti con normali livelli di GH, mentre per i pazienti che hanno un deficit di GH, il trattamento standard con ormone della crescita è appropriato.
La prognosi della sindrome di Rothmund-Thomson è variabile
L’attesa di vita è normale in assenza di tumori, mentre la prognosi dei pazienti con tumore dipende dalla qualità e dalla frequenza dello screening tumorale e dalla precocità del trattamento.
I pazienti affetti da sindrome di Rothmund-Thomson possono essere particolarmente sensibili agli effetti collaterali della chemioterapia e presentano un rischio elevato di sviluppare tumori secondari (rischio del 5% di sviluppare un tumore della cute).
Se il trattamento è adeguato, la prognosi delle forme lievi della malattia è favorevole e l’aspettativa di vita è approssimativamente sovrapponibile a quella della popolazione generale.
Per approfondire
Emergency Live ancora più…live: scarica la nuova app gratuita del tuo giornale per iOS e Android
Malattie rare: l’iperinsulinismo congenito
Malattie rare: la Sindrome di Von Hippel-Lindau
Malattie rare: l’interstiziopatia polmonare causa il 35% di decessi dei pazienti con sclerodermia
Sindrome Guillain-Barrè, il neurologo: “Nessun nesso con Covid o vaccino”
Formazione nel soccorso, Sindrome Neurolettica Maligna: cos’è e come si affronta
Pemfigo: scopriamo questa rara malattia autoimmune della pelle
Malattie autoimmuni: la sabbia negli occhi della Sindrome di Sjögren
Cos’è l’impetigine in adulti e bambini e come curarsi
Malattie rare: la Displasia Setto-Ottica
Progeria: che cos’è, sintomi, cause, diagnosi e possibili cure
Malformazioni del piede: il metatarso addotto o metatarso varo