
Sindrome del QT lungo: cause, diagnosi, valori, cura, farmaci
Con sindrome del QT lungo (LQTS) si indica un insieme di sintomi determinati da una anomalia cardiaca caratterizzata da una ritardata ripolarizzazione delle cellule miocardiche ed associata a sincope (svenimento con perdita di coscienza e tono posturale)
La sincope è determinata il più delle volte da aritmie maligne, specialmente torsioni di punta, che possono degenerare in fibrillazione ventricolare, sino all’arresto cardiaco irreversibile del soggetto colpito (morte cardiaca improvvisa).
Le aritmie nei pazienti affetti da LQTS sono spesso scatenate dall’esercizio fisico o da stimoli emotivi molto forti, come il terrore.
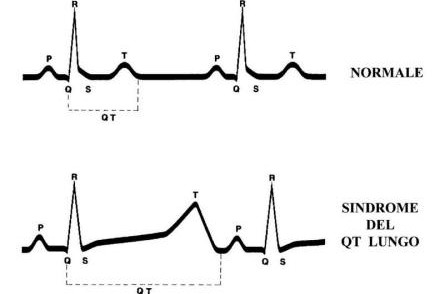
Gli individui portatori di LQTS hanno un caratteristico prolungamento dell’intervallo QT all’elettrocardiogramma: tale intervallo si misura dall’inizio dell’onda Q al termine dell’onda T (vedi immagine in alto).
Alla base di tutte le forme di LQTS c’è una anormale ripolarizzazione del miocardio.
Le anomalie della ripolarizzazione causano differenze nella refrattarietà dei miocardiociti.
A causa di queste differenze, eventuali post-depolarizzazioni (che si verificano più di frequente nei pazienti affetti da LQTS) possono propagarsi alle cellule contigue, conducendo ad aritmie ventricolari da rientro.
![]() Epidemiologia e fattori di rischio nella sindrome del QT Lungo (LQTS)
Epidemiologia e fattori di rischio nella sindrome del QT Lungo (LQTS)

Nei soggetti geneticamente predisposti, costituiscono fattori di rischio per l’insorgenza di aritmie maligne gli incrementi improvvisi del tono simpatico, come può accadere in concomitanza di sforzi eccessivi o emozioni violente.
La sindrome colpisce prevalentemente le giovani donne. Fra i soggetti affetti, l’incidenza di torsione di punta, sincope e morte cardiaca improvvisa è più elevata in caso di sordità congenita, precedente tachiaritmia o sincope; la stessa aumenta inoltre proporzionalmente all’allungamento dell’intervallo QT.
Si calcola che il rischio relativo di aritmie maligne aumenti di 1,1 – 1,2 volte per ciascun prolungamento di 10 msec del QTc oltre i valori normali.
Varianti cliniche
Si distinguono due diverse sindromi congenite caratterizzate da allungamento dell’intervallo QT e rischio di morte improvvisa per aritmie ventricolari:
- Sindrome di Romano-Ward, che viene ereditata secondo il modello autosomico dominante (non associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza).
- Sindrome di Jervell-Lange-Nielsen, che viene ereditata secondo il modello autosomico recessivo (associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza).
Cause della LQTS
Nella LQTS si distinguono due forme: congenita (più rara) ed acquisita (più frequente).
Forme acquisite
La maggior parte dei casi riscontrati nella pratica clinica riguarda forme acquisite, che possono essere suddivise in due categorie: quelle generate da disturbi dell’equilibrio idro-elettrolitico e quelle dipendenti dalla somministrazione di farmaci.
Forme indotte da disordini elettrolitici:
- ipokaliemia
- ipomagnesiemia
- ipocalcemia
Forme indotte da farmaci:
- farmaci antiaritmici
- Chinidina
- Amiodarone
- Sotalolo
- Procainamide
- Ranolazina
- Anti-istaminici
- terfenadina
- astemizolo
- mizolastina
- Antibiotici Macrolidi
- Eritromicina
- Alcuni antibiotici fluorochinolonici
- Ansiolitici maggiori
- Antidepressivi triciclici
- Agenti attivi sulla motilità gastro-intestinale
- Cisapride
- Domperidone
- farmaci antipsicotici
- Aloperidolo
- Quetiapina
- Tioridazina
- Droperidolo
- Analgesici
- Metadone
- LAAM
Proprio come le forme congenite di LQTS, le forme acquisite possono condurre ad aritmie mortali.
Il trattamento consiste nella correzione dello squilibrio elettrolitico, nella risoluzione della sua causa e nella sospensione della terapia con il farmaco indiziato dell’allungamento del QT.
Dato il largo impiego, la tendenza ad interazioni con altri farmaci e la insita capacità di determinare allungamento dell’intervallo QT, l’eritromicina è probabilmente la causa prevalente di sindrome acquisita del QT lungo.
Infatti, l’uso di eritromicina è associato con un’incidenza di morte improvvisa più che doppia rispetto ad altri antibiotici.
In aggiunta alle due maggiori categorie elencate sopra, deve essere ricordato che esistono altre cause di prolungamento dell’intervallo QT come l’anoressia nervosa, l’ipotiroidismo, l’infezione da HIV, la miocardite e l’infarto miocardico.
Forme congenite
Le forme congenite di LQTS possono essere determinate da mutazioni di uno di diversi geni finora identificati.
Queste mutazioni tendono a prolungare la durata del potenziale d’azione ventricolare (APD), allungando in tal modo l’intervallo QT.
Le forme congenite possono essere ereditate come carattere autosomico dominante o autosomico recessivo.
La forma autosomica recessiva è associata ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza (LQTS8) o a sordità neurologica congenita (LQTS1).
Un numero sempre crescente di loci genici viene identificato in associazione alle LQTS. I test genetici per le LQTS è disponibile nella pratica clinica e può essere di aiuto anche per impostare la terapia appropriata (Overview of LQTS Genetic Testing).
Le più comuni cause di LQTS riguardano mutazioni nei geni KCNQ1 (LQTS1), KCNH2 (LQTS2) e SCN5A (LQT3).
Sindrome di Jervell e Lange-Nielsen
È la forma autosomica recessiva di LQTS.
È associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza.
Essa è causata specificatamente da una mutazione dei geni KCNE1 e KCNQ1.
Degli individui non trattati, circa il 50% muore entro l’età di 15 anni a causa di aritmie ventricolari.
Sindrome di Romano-Ward
La sindrome di Romano-Ward è la forma autosomica dominante di LQTS.
Non è associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza.
Diagnosi e valori della Sindrome del QT Lungo:
La diagnosi di LQTS spesso non è facile dal momento che il 2.5% della popolazione sana ha un QT prolungato, e il 10–15% dei pazienti affetti da LQTS ha un intervallo QT normale: spesso perfino alcuni atleti professionisti possono averla senza che il personale medico se ne accorga.
Un criterio diagnostico abitualmente usato è si basa sul LQTS “diagnostic score”.

Il punteggio è calcolato assegnando punti in base a vari criteri elencati qui di seguito.
Con 4 o più punti la probabilità di LQTS è alta, e con 1 punto o meno la probabilità è bassa; 2 o 3 punti indicano una probabilità intermedia.
QTc (definito come intervallo QT/radice quadrata dell’intervallo RR)
- >= 480 msec – 3 punti
- 460-470 msec – 2 punti
- 450 msec e genere maschile – 1 punto
- Tachicardie ventricolari tipo torsades de pointes – 2 punti
- Alternanza dell’onda T – 1 punto
- Avvallamento dell’onda T in almeno 3 derivazioni all’ECG- 1 punto
- Bassa frequenza cardiaca per l’età (bambini) – 0.5 punti
- Sincope (non possono essere assegnati punti sia alla sincope che alle torsioni di punta allo stesso soggetto)
- In caso di stress – 2 punti
- Al di fuori di condizioni di stress – 1 punto
- Sordità congenita – 0.5 punti
- Storia familiare (lo stesso membro della famiglia non può essere conteggiato sia per la morte improvvisa sia per la LQTS)
- Altri membri della famiglia con diagnosi sicura di LQTS – 1 punto
- Morte improvvisa nei familiari stretti (membri con età inferiore a 30 anni) – 0.5 punti
Terapia
Nei pazienti asintomatici, senza dimostrazione di aritmie ventricolari ed in assenza di anamnesi familiare positiva per morte improvvisa, è raccomandata la sola osservazione ed eventualmente la terapia farmacologica senza necessità di arrivare al dosaggio massimo tollerato.
Nelle LQTS1 e LQTS2 possono essere utilizzati i betabloccanti; mentre nelle LQTS 3 sono preferibili gli anti-aritmici di classe Ib, come la mexiletina.
Gli stessi farmaci possono essere adoperati nel trattamento dei pazienti in urgenza, con l’accortezza di utilizzare soltanto la lidocaina fin quando non venga confermata la diagnosi di LQTS1 o LQTS2, poiché i betabloccanti possono peggiorare le aritmie nei pazienti portatori di LQTS3.
La terapia anti-aritmica deve essere, invece, adottata e condotta al dosaggio massimo tollerato in quei paziente asintomatici ma con evidenza di aritmie ventricolari non sostenute e storia familiare di morte improvvisa.
In questi pazienti non è strettamente raccomandato l’impianto del defibrillatore cardiaco impiantabile (ICD).
Quest’ultimo è, invece, assolutamente indicato (indicazione di classe I) nei pazienti con sincope o che abbiano già avuto un episodio di arresto cardiaco.
La funzione di pace-maker del dispositivo deve essere sfruttata in coloro che presentano aritmie in occasione di bradicardia o pause del ritmo cardiaco.
Nei pazienti che presentino ancora dei sintomi, nonostante la terapia medica ottimale, è indicato l’intervento chirurgico di gangliectomia cervico-toracica sinistra, con distruzione del ganglio stellato e dei primi tre o quattro gangli simpatici toracici.
Prognosi e rischio
Per i pazienti affetti da LQTS e non trattati il rischio di incorrere in un evento (sincope o arresto cardiaco) può essere stimato conoscendo il loro genotipo (LQTS1-10), dal genere e dall’intervallo QTc.
Rischio elevato (>50%)
QTc>500 msec LQTS1 & LQTS2 & LQTS3 (maschi)
Rischio intermedio (30-50%)
QTc>500 msec LQTS3 (femmine)
QTc<500 msec LQTS2 (femmine)& LQTS3
Rischio basso (<30%)
QTc<500 msec LQT1 & LQT2 (maschi)
Sindrome del QT lungo e sport
I pazienti affetti da LQTS, sotto stretto controllo cardiologico, possono comunque svolgere sport, evitando però quelli che includono sforzi elevati per tempi prolungati e gli sport acquatici come il nuoto e immersioni.
Per approfondire:
Emergency Live ancora più…live: scarica la nuova app gratuita del tuo giornale per iOS e Android
La sindrome del cuore spezzato è in aumento: conosciamo la cardiomiopatia di Takotsubo
Defibrillatore: cos’è, come funziona, prezzo, voltaggio, manuale ed esterno
La corretta manutenzione del defibrillatore per garantirne la massima efficienza
Manovra di Heimlich: scopriamo assieme cos’è e come praticarla
Supporto vitale di base (BTLS) e avanzato (ALS) al paziente traumatizzato
Procedure sul paziente: che cosa si intende per cardioversione elettrica esterna?
Massaggio cardiaco: quante compressioni al minuto?
Sincope cardiaca: che cos’è, come si diagnostica e chi colpisce
Holter cardiaco: come funziona e quando serve?
Sepsi, ecco perché un’infezione è un pericolo e una minaccia per il cuore
L’importanza della Rianimazione Cardiopolmonare: intervenire tempestivamente può salvare una vita
Differenza tra cardioversione spontanea, elettrica e farmacologica
Che cos’è un Cardiovertitore? Panoramica sul defibrillatore impiantabile
Che cos’è la cardiomiopatia di takotsubo (o sindrome del cuore spezzato)?
L’ECG del paziente: come leggere un elettrocardiogramma in modo semplice
La regola del 1/2 RR nella valutazione del QT