

Langt QT-syndrom: årsaker, diagnose, verdier, behandling, medisinering
Langt QT-syndrom (LQTS) refererer til et sett med symptomer forårsaket av en hjerteabnormitet preget av forsinket repolarisering av myokardceller og assosiert med synkope (besvimelse med tap av bevissthet og postural tonus)
Synkope er oftest forårsaket av ondartede arytmier, spesielt tipptorsjoner, som kan utarte seg til ventrikkelflimmer, noe som fører til irreversibel hjertestans hos den berørte personen (plutselig hjertedød).
Arytmier hos LQTS-pasienter utløses ofte av trening eller svært sterke emosjonelle stimuli, som for eksempel terror.
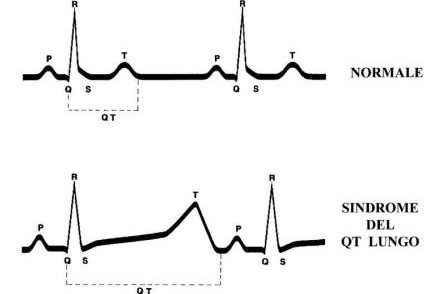
Personer med LQTS har en karakteristisk forlengelse av QT-intervallet på elektrokardiogrammet: dette intervallet måles fra begynnelsen av Q-bølgen til slutten av T-bølgen (se bildet over).
Under alle former for LQTS ligger en unormal repolarisering av myokardiet.
Abnormiteter i repolarisering forårsaker forskjeller i myokard refraktæritet.
På grunn av disse forskjellene kan eventuelle etterdepolarisasjoner (som forekommer hyppigere hos LQTS-pasienter) forplante seg til tilstøtende celler, noe som fører til re-entry ventrikulære arytmier.
Epidemiologi og risikofaktorer ved lang QT-syndrom (LQTS)
Hos genetisk disponerte individer er plutselige økninger i sympatisk tonus, slik som kan oppstå ved overdreven anstrengelse eller voldsomme følelser, risikofaktorer for utbruddet av ondartede arytmier.
Syndromet rammer hovedsakelig unge kvinner. Blant de berørte er forekomsten av tipptorsjon, synkope og plutselig hjertedød høyere ved medfødt døvhet, tidligere takyarytmi eller synkope; det øker også proporsjonalt med forlengelsen av QT-intervallet.
Den relative risikoen for ondartede arytmier er estimert til å øke 1.1 – 1.2 ganger for hver 10 msek forlengelse av QTc-intervallet utover normale verdier.
Kliniske varianter
Vi skiller mellom to forskjellige medfødte syndromer preget av forlengelse av QT-intervallet og risiko for plutselig død på grunn av ventrikulære arytmier:
- Romano-Ward syndrom, som er arvet i henhold til den autosomal dominante modellen (ikke assosiert med medfødt nevrologisk døvhet eller andre medfødte hjertesykdommer, autisme, fullstendig syndaktyli og immunsvikt).
- Jervell-Lange-Nielsen syndrom, som er arvet i det autosomale recessive mønsteret (assosiert med medfødt nevrologisk døvhet eller andre medfødte hjertesykdommer, autisme, fullstendig syndaktyli og immunsvikt).
Årsaker til LQTS
Det er to former for LQTS: medfødt (sjeldnere) og ervervet (hyppigere).
Innhentede former
De fleste tilfeller sett i klinisk praksis involverer ervervede former, som kan deles inn i to kategorier: de som genereres av forstyrrelser i hydroelektrolyttbalansen og de som er avhengige av medikamentadministrasjon.
Former indusert av elektrolyttforstyrrelser
- hypokalemi
- hypomagnesemi
- hypokalsemi
- Legemiddelinduserte former
- antiarytmika
- Kinidin
- Amiodarone
- Sotalol
- Prokainamid
- ranolazin
- Anti-histaminer
- terfenadin
- astemizol
- mizolastin
- Makrolide antibiotika
- erytromycin
- Visse fluorokinolon-antibiotika
- Store anxiolytika
- Trisykliske antidepressiva
- Midler som er aktive på gastrointestinal motilitet
- cisaprid
- Domperidone
- Antipsykotiske legemidler
- Haloperidol
- Quetiapine
- Tioridazin
- droperidol
- analgetika
- Metadon
- LAAM
Akkurat som medfødte former for LQTS, kan ervervede former føre til livstruende arytmier.
Behandlingen består i å korrigere elektrolyttubalansen, løse årsaken og avbryte behandlingen med legemidlet som indikerer QT-forlengelse.
Gitt sin brede bruk, tendens til interaksjoner med andre legemidler og iboende evne til å forårsake QT-intervallforlengelse, er erytromycin sannsynligvis den dominerende årsaken til ervervet lang QT-syndrom.
Faktisk er erytromycinbruk assosiert med en forekomst av plutselig død mer enn det dobbelte av andre antibiotika.
I tillegg til de to hovedkategoriene som er oppført ovenfor, må det huskes at det er andre årsaker til QT-intervallforlengelse som anorexia nervosa, hypotyreose, HIV-infeksjon, myokarditt og hjerteinfarkt.
Medfødte former
Medfødte former for LQTS kan bestemmes av mutasjoner i ett av flere gener identifisert til dags dato.
Disse mutasjonene har en tendens til å forlenge varigheten av det ventrikulære handlingspotensialet (APD), og dermed forlenge QT-intervallet.
De medfødte formene kan arves som en autosomal dominant eller autosomal recessiv karakter.
Den autosomale recessive formen er assosiert med andre medfødte hjertesykdommer, autisme, fullstendig syndaktyli og immunsvikt (LQTS8) eller med medfødt nevrologisk døvhet (LQTS1).
Et økende antall gen-loci blir identifisert i forbindelse med LQTS. Genetisk testing for LQTS er tilgjengelig i klinisk praksis og kan også hjelpe med å sette riktig terapi (Oversikt over LQTS genetisk testing).
De vanligste årsakene til LQTS involverer mutasjoner i genene KCNQ1 (LQTS1), KCNH2 (LQTS2) og SCN5A (LQT3).
VERDENS LEDENDE SELSKAP FOR DEFIBRILLATORER OG NØDSIDSINSKE ENHETER'? BESØK ZOLL-BODEN PÅ NØD-EXPO
Jervell og Lange-Nielsen syndrom
Det er den autosomale recessive formen av LQTS.
Det er assosiert med medfødt nevrologisk døvhet eller andre medfødte hjertesykdommer, autisme, fullstendig syndaktyli og immunsvikt.
Det er spesielt forårsaket av en mutasjon i KCNE1- og KCNQ1-genene.
Av ubehandlede individer dør omtrent 50 % ved fylte 15 år på grunn av ventrikulære arytmier.
Romano-Ward syndrom
Romano-Ward syndrom er den autosomalt dominerende formen for LQTS.
Det er ikke assosiert med medfødt nevrologisk døvhet eller andre medfødte hjertesykdommer, autisme, fullstendig syndaktyli og immunsvikt.
Diagnose og verdier av langt QT-syndrom:
Diagnosen LQTS er ofte ikke lett siden 2.5 % av den friske befolkningen har en forlenget QT, og 10-15 % av LQTS-pasientene har normalt QT-intervall: ofte kan til og med noen profesjonelle idrettsutøvere ha det uten at medisinsk personell merker det.
Et ofte brukt diagnostisk kriterium er basert på LQTS 'diagnostisk poengsum'.

Poengsummen beregnes ved å tildele poeng i henhold til ulike kriterier oppført nedenfor.
Med 4 eller flere poeng er sannsynligheten for LQTS høy, og med 1 poeng eller mindre er sannsynligheten lav; 2 eller 3 poeng indikerer en middels sannsynlighet.
QTc (definert som QT-intervall/kvadratrot av RR-intervall)
- >= 480 msek – 3 poeng
- 460-470 msek – 2 poeng
- 450 msek og mannlig kjønn – 1 poeng
- Ventrikulære torsades de pointes type takykardier – 2 poeng
- T-bølgeveksling – 1 poeng
- T-bølgeskred i minst 3 avledninger på EKG – 1 poeng
- Lav puls for alder (barn) – 0.5 poeng
- Synkope (ingen poeng kan gis for både synkope og topptorsjon i samme fag)
- Under stressforhold – 2 poeng
- Utenfor stressforhold – 1 poeng
- Medfødt døvhet – 0.5 poeng
- Familiehistorie (det samme familiemedlemmet kan ikke telles for både plutselig død og LQTS)
- Andre familiemedlemmer med sikker diagnose LQTS – 1 poeng
- Plutselig død hos nære familiemedlemmer (medlemmer under 30 år) – 0.5 poeng
Terapi
Hos asymptomatiske pasienter uten påvisning av ventrikulære arytmier og i fravær av en positiv familiehistorie med plutselig død, anbefales observasjon alene og muligens medikamentell behandling uten behov for å gå til den maksimalt tolererte dosen.
I LQTS1 og LQTS2 kan betablokkere brukes; i LQTS 3 er klasse Ib antiarytmika som mexiletin å foretrekke.
De samme legemidlene kan brukes til å behandle akutte pasienter, med forbehold om at kun lidokain skal brukes inntil diagnosen LQTS1 eller LQTS2 er bekreftet, da betablokkere kan forverre arytmier hos LQTS3-pasienter.
Antiarytmisk terapi bør imidlertid tas i bruk og utføres med den maksimalt tolererte dosen hos de pasientene som er asymptomatiske, men som har tegn på ikke-vedvarende ventrikulære arytmier og en familiehistorie med plutselig død.
Implanterbar cardioverter Defibrillator (ICD) implantasjon er ikke strengt anbefalt hos disse pasientene.
Sistnevnte er imidlertid absolutt indisert (klasse I indikasjon) hos pasienter med synkope eller som allerede har hatt en episode med hjertestans.
Enhetens pacemakerfunksjon må utnyttes hos de som har arytmier under bradykardi eller pauser i hjerterytmen.
Hos pasienter som fortsatt viser symptomer til tross for optimal medisinsk terapi, er venstre cervikal-thorax gangliektomi kirurgi indisert, med ødeleggelse av stellate ganglion og de første tre eller fire thorax sympatiske ganglier.
Prognose og risiko
For ubehandlede LQTS-pasienter kan risikoen for en hendelse (synkope eller hjertestans) estimeres ved å kjenne deres genotype (LQTS1-10), kjønn og QTc-intervall.
Høy risiko (>50 %)
QTc>500 msek LQTS1 & LQTS2 & LQTS3 (hanner)
Middels risiko (30-50 %)
QTc>500 msek LQTS3 (kvinner)
QTc<500 msek LQTS2 (kvinner) og LQTS3
Lav risiko (<30 %)
QTc<500 msek LQT1 og LQT2 (hanner)
Langt QT-syndrom og sport
Pasienter med LQTS, under streng kardiologisk kontroll, kan fortsatt drive med sport, men unngå de som inkluderer langvarig høy anstrengelse og vannsport som svømming og dykking.
Les også:
Emergency Live enda mer...Live: Last ned den nye gratisappen til avisen din for iOS og Android
Hjertesykdom: Hva er kardiomyopati?
Inflammasjoner i hjertet: Myokarditt, infeksjonsendokarditt og perikarditt
Hjertemurl: Hva det er og når det skal bekymres
Broken Heart Syndrome er på vei oppover: Vi kjenner Takotsubo kardiomyopati
Hva er en cardioverter? Oversikt over implanterbar defibrillator
Squicciarini Rescue Velger Emergency Expo: American Heart Association BLSD- og PBLSD-opplæringskurs
'D' For Deads, 'C' For Cardioversion! - Defibrillering og flimmer hos pediatriske pasienter
Hjertebetennelser: Hva er årsakene til perikarditt?
Har du episoder med plutselig takykardi? Du kan lide av Wolff-Parkinson-White syndrom (WPW)
Å vite at trombose kan gripe inn på blodproppen
Pasientprosedyrer: Hva er ekstern elektrisk kardioversjon?
Øke arbeidsstyrken til EMS, trene lekfolk i bruk av AED
Forskjellen mellom spontan, elektrisk og farmakologisk kardioversjon
Hva er Takotsubo Kardiomyopati (Broken Heart Syndrome)?
Pasientens EKG: Hvordan lese et elektrokardiogram på en enkel måte
Stressøvelsestest som induserer ventrikulære arytmier hos personer med LQT -intervall