

Långt QT-syndrom: orsaker, diagnos, värden, behandling, medicinering
Långt QT-syndrom (LQTS) hänvisar till en uppsättning symtom som orsakas av en hjärtabnormalitet som kännetecknas av fördröjd repolarisering av myokardceller och förknippas med synkope (svimning med förlust av medvetande och postural tonus)
Synkope orsakas oftast av maligna arytmier, särskilt spetstorsioner, som kan degenerera till kammarflimmer, vilket leder till irreversibelt hjärtstillestånd hos den drabbade personen (plötslig hjärtdöd).
Arytmier hos LQTS-patienter utlöses ofta av träning eller mycket starka känslomässiga stimuli, såsom terror.
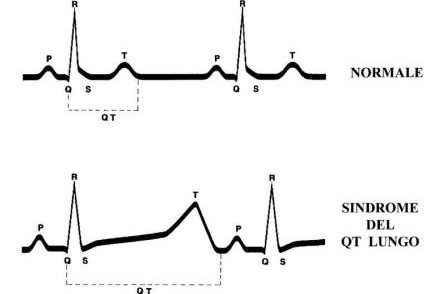
Individer med LQTS har en karakteristisk förlängning av QT-intervallet på elektrokardiogrammet: detta intervall mäts från början av Q-vågen till slutet av T-vågen (se bilden ovan).
Bakom alla former av LQTS ligger en onormal repolarisering av myokardiet.
Abnormaliteter i repolarisering orsakar skillnader i myokardial refraktäritet.
På grund av dessa skillnader kan eventuella efterdepolarisationer (som förekommer oftare hos LQTS-patienter) fortplanta sig till intilliggande celler, vilket leder till ventrikulära arytmier som återkommer.
Epidemiologi och riskfaktorer vid långt QT-syndrom (LQTS)
Hos genetiskt predisponerade individer är plötsliga ökningar av sympatisk tonus, som kan uppstå vid överdriven ansträngning eller våldsamma känslor, riskfaktorer för uppkomsten av maligna arytmier.
Syndromet drabbar främst unga kvinnor. Bland de drabbade är förekomsten av spetstorsion, synkope och plötslig hjärtdöd högre vid medfödd dövhet, tidigare takyarytmi eller synkope; det ökar också i proportion till förlängningen av QT-intervallet.
Den relativa risken för maligna arytmier uppskattas öka 1.1 – 1.2 gånger för varje 10 msek förlängning av QTc-intervallet utöver normala värden.
Kliniska varianter
Vi särskiljer två olika medfödda syndrom som kännetecknas av förlängning av QT-intervallet och risk för plötslig död på grund av ventrikulära arytmier:
- Romano-Wards syndrom, som ärvs enligt den autosomala dominanta modellen (ej förknippat med medfödd neurologisk dövhet eller andra medfödda hjärtsjukdomar, autism, fullständig syndaktyli och immunbrist).
- Jervell-Lange-Nielsens syndrom, som ärvs i det autosomala recessiva mönstret (associerat med medfödd neurologisk dövhet eller andra medfödda hjärtsjukdomar, autism, fullständig syndaktyli och immunbrist).
Orsaker till LQTS
Det finns två former av LQTS: medfödd (sällsyntare) och förvärvad (mer frekvent).
Förvärvade former
De flesta fall som ses i klinisk praxis involverar förvärvade former, som kan delas in i två kategorier: de som genereras av störningar i hydroelektrolytbalansen och de som är beroende av läkemedelsadministrering.
Former inducerade av elektrolytrubbningar
- hypokalemi
- hypomagnesemi
- hypokalcemi
- Läkemedelsinducerade former
- antiarytmika
- Kinidin
- Amiodaron
- Sotalol
- Prokainamid
- Ranolazin
- Antihistaminer
- terfenadin
- astemizol
- mizolastin
- Makrolidantibiotika
- erytromycin
- Vissa fluorokinolonantibiotika
- Större anxiolytika
- Tricykliska antidepressiva
- Medel aktiva på gastrointestinala motilitet
- Cisaprid
- Domperidon
- Antipsykotiska läkemedel
- Haloperidol
- Quetiapin
- Tioridazin
- Droperidol
- Smärtstillande medel
- Metadon
- LAM
Precis som medfödda former av LQTS kan förvärvade former leda till livshotande arytmier.
Behandlingen består av att korrigera elektrolytobalansen, lösa dess orsak och att avbryta behandlingen med läkemedlet som tyder på QT-förlängning.
Med tanke på dess breda användning, tendens till interaktioner med andra läkemedel och inneboende förmåga att orsaka förlängning av QT-intervallet, är erytromycin förmodligen den dominerande orsaken till förvärvat långt QT-syndrom.
Faktum är att användning av erytromycin är associerad med en incidens av plötslig död mer än dubbelt så stor som andra antibiotika.
Utöver de två huvudkategorierna som anges ovan måste man komma ihåg att det finns andra orsaker till QT-intervallförlängning såsom anorexia nervosa, hypotyreos, HIV-infektion, myokardit och hjärtinfarkt.
Medfödda former
Medfödda former av LQTS kan bestämmas genom mutationer i en av flera gener som hittills identifierats.
Dessa mutationer tenderar att förlänga varaktigheten av den ventrikulära aktionspotentialen (APD), vilket förlänger QT-intervallet.
De medfödda formerna kan ärvas som en autosomal dominant eller autosomal recessiv karaktär.
Den autosomala recessiva formen är associerad med andra medfödda hjärtsjukdomar, autism, fullständig syndaktyli och immunbrist (LQTS8) eller med medfödd neurologisk dövhet (LQTS1).
Ett ökande antal genloci identifieras i samband med LQTS. Genetisk testning för LQTS är tillgänglig i klinisk praxis och kan också hjälpa till att ställa in lämplig terapi (Översikt över LQTS genetisk testning).
De vanligaste orsakerna till LQTS involverar mutationer i generna KCNQ1 (LQTS1), KCNH2 (LQTS2) och SCN5A (LQT3).
Jervells och Lange-Nielsens syndrom
Det är den autosomalt recessiva formen av LQTS.
Det är förknippat med medfödd neurologisk dövhet eller andra medfödda hjärtsjukdomar, autism, fullständig syndaktyli och immunbrist.
Det är specifikt orsakat av en mutation i generna KCNE1 och KCNQ1.
Av obehandlade individer dör cirka 50 % vid 15 års ålder på grund av ventrikulära arytmier.
Romano-Wards syndrom
Romano-Wards syndrom är den autosomalt dominerande formen av LQTS.
Det är inte associerat med medfödd neurologisk dövhet eller andra medfödda hjärtsjukdomar, autism, fullständig syndaktyli och immunbrist.
Diagnos och värden av Long QT Syndrome:
Diagnosen av LQTS är ofta inte lätt eftersom 2.5% av den friska befolkningen har en förlängd QT, och 10-15% av LQTS-patienterna har ett normalt QT-intervall: ofta kan även vissa professionella idrottare ha det utan att medicinsk personal märker det.
Ett vanligt använt diagnostiskt kriterium är baserat på LQTS 'diagnostiska poäng'.

Poängen beräknas genom att tilldela poäng enligt olika kriterier som anges nedan.
Med 4 eller fler poäng är sannolikheten för LQTS hög, och med 1 poäng eller mindre är sannolikheten låg; 2 eller 3 poäng indikerar en medelsannolikhet.
QTc (definieras som QT-intervall/kvadratrot av RR-intervall)
- >= 480 msek – 3 poäng
- 460-470 msek – 2 poäng
- 450 msek och manligt kön – 1 poäng
- Ventrikulära torsades de pointes typ takykardier – 2 poäng
- T-vågsväxling – 1 poäng
- T-våg lavin i minst 3 avledningar på EKG – 1 poäng
- Låg puls för ålder (barn) – 0.5 poäng
- Synkope (inga poäng kan ges för både synkope och peak torsion i samma ämne)
- Under stressförhållanden – 2 poäng
- Utanför stressförhållanden – 1 poäng
- Medfödd dövhet – 0.5 poäng
- Familjehistoria (samma familjemedlem kan inte räknas för både plötslig död och LQTS)
- Övriga familjemedlemmar med säker diagnos LQTS – 1 poäng
- Plötsligt dödsfall hos nära familjemedlemmar (medlemmar under 30 år) – 0.5 poäng
Terapi
Hos asymtomatiska patienter utan påvisande av ventrikulära arytmier och i avsaknad av positiv familjehistoria av plötslig död, rekommenderas enbart observation och eventuellt läkemedelsbehandling utan att behöva gå till den maximalt tolererade dosen.
I LQTS1 och LQTS2 kan betablockerare användas; i LQTS 3 är klass Ib antiarytmika som mexiletin att föredra.
Samma läkemedel kan användas för att behandla akutpatienter, med förbehållet att endast lidokain ska användas tills diagnosen LQTS1 eller LQTS2 är bekräftad, eftersom betablockerare kan förvärra arytmier hos LQTS3-patienter.
Antiarytmisk terapi bör dock användas och utföras med den maximalt tolererade dosen hos de patienter som är asymtomatiska men som har tecken på icke ihållande ventrikulära arytmier och en familjehistoria med plötslig död.
Implanterbar cardioverter defibrillatorn (ICD) implantation rekommenderas inte strikt för dessa patienter.
Det senare är dock absolut indicerat (klass I-indikation) hos patienter med synkope eller som redan har haft en episod av hjärtstopp.
Enhetens pacemakerfunktion måste utnyttjas hos dem som uppvisar arytmier under bradykardi eller pauser i hjärtrytmen.
Hos patienter som fortfarande uppvisar symtom trots optimal medicinsk terapi, är vänster cervikal-thorax gangliektomi operation indicerad, med förstörelse av stella gangliet och de första tre eller fyra thorax sympatiska ganglierna.
Prognos och risk
För obehandlade LQTS-patienter kan risken för en händelse (synkope eller hjärtstillestånd) uppskattas genom att känna till deras genotyp (LQTS1-10), kön och QTc-intervall.
Hög risk (>50 %)
QTc>500 msek LQTS1 & LQTS2 & LQTS3 (hanar)
Mellanliggande risk (30-50 %)
QTc>500 msek LQTS3 (honor)
QTc<500 msek LQTS2 (honor)& LQTS3
Låg risk (<30 %)
QTc<500 ms LQT1 & LQT2 (hanar)
Långt QT-syndrom och sport
Patienter med LQTS, under strikt kardiologisk kontroll, kan fortfarande utöva sport, men undvik sådana som inkluderar långvarig hög ansträngning och vattensporter som simning och dykning.
Läs också:
Emergency Live Ännu mer...Live: Ladda ner den nya gratisappen för din tidning för IOS och Android
Hjärtsjukdom: Vad är kardiomyopati?
Hjärtinflammationer: Myokardit, infektiös endokardit och perikardit
Hjärtmummor: Vad det är och när man ska oroa sig
Trasigt hjärtsyndrom är på uppgång: Vi känner till Takotsubo kardiomyopati
Vad är en cardioverter? Översikt över implanterbar defibrillator
'D' för döda, 'C' för kardioversion! - Defibrillering och flimmer hos barn
Inflammationer i hjärtat: Vad är orsakerna till perikardit?
Har du episoder av plötslig takykardi? Du kan lida av Wolff-Parkinson-White syndrom (WPW)
Att veta Trombos Att Ingripa På Blodpropp
Patientprocedurer: Vad är extern elektrisk elkonvertering?
Öka personalstyrkan hos EMS, utbilda lekmän i att använda AED
Skillnaden mellan spontan, elektrisk och farmakologisk elkonvertering
Vad är Takotsubo Kardiomyopati (Broken Heart Syndrome)?
Patientens EKG: Hur man läser ett elektrokardiogram på ett enkelt sätt
Stressövningstest som inducerar ventrikulära arytmier hos personer med LQT -intervall